

Developing a new drug is a challenging task as it requires to go through a multi-step process. From developing a target discovery to its clinical trials, this process might take a considerable amount of time. And after going through the whole process, only a few of the drugs actually reach the market. But thanks to Artificial Intelligence & Machine Learning that has made this task much easier.
Till now, GAN was mostly used to generate images, but now we can use these AI models to search from 106 chemical compounds and try to generate the leading molecule for the target drug. One of the methods that is generated by the researchers of Pennsylvania State University is via Quantum GAN. The paper Quantum Generative Models for Small Molecule Drug Discovery was first submitted to ArXiv in January 2021 by Junde Li, Rasit Topaloglu, Swaroop Ghosh.
This model proposes a qubit-efficient Quantum GAN with Hybrid Generator(QGAN-HG) to learn a huge amount of representation of molecules by searching 106 large chemical spaces with few qubits. QGAN-HG gives better results than classical GAN.
If you are reading the article, we assume that you are already aware of the GAN model. If not, you can check this archive to know more about it. Let’s get started with some quantum related terminologies which help to understand Quantum Neural Network.
Quantum Circuits
Quantum Circuits is the ordered-collection of gates that changes the qubits’ state to perform some quantum operations.
Quantum noise
A quantum noise can be described as anything that has malfunctioned the quantum computer. When qubits are exposed to any kind of source that causes noise, quantum computers’ information gets degraded.
In quantum GAN, GAN model’s generators and discriminators are trained on quantum computers or devices which can easily process high-dimensional data(or quantum data).
Quantum GAN with Hybrid Generator
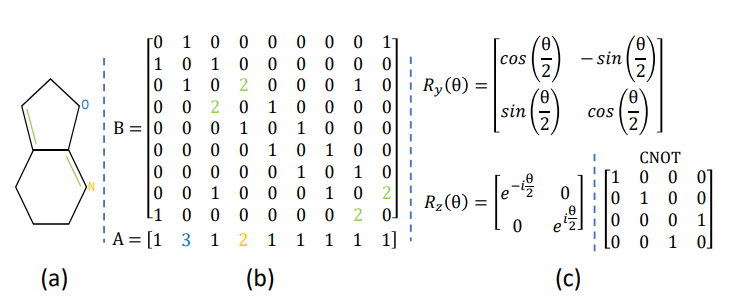
QGAN-HG consists of parameterized quantum circuits that give a feature vector of qubit size dimension. A classic neural network is required to yield the required atom vector and bond matrix to represent the molecule in a graph where nodes signify the atoms and edges denote the bonds.
QGAN-HG Quantum Circuit
This circuit gives the feature vector with the help of a quantum layer that does the computation in O(poly(log(M))) time. The quantum layer consists of three parts: initialization, parameterized and measurement stages. In the initialization section: two parameters z1 and z2 are sampled uniformly from[−π, π]. These two parameters are then converted into a mathematical form and repeated for all layers to generate a unitary matrix U(θ). Then the feature vector is obtained by applying quantum operations on the final quantum state. Below in the figure mentioned Ry and Rz are the rotation gates.
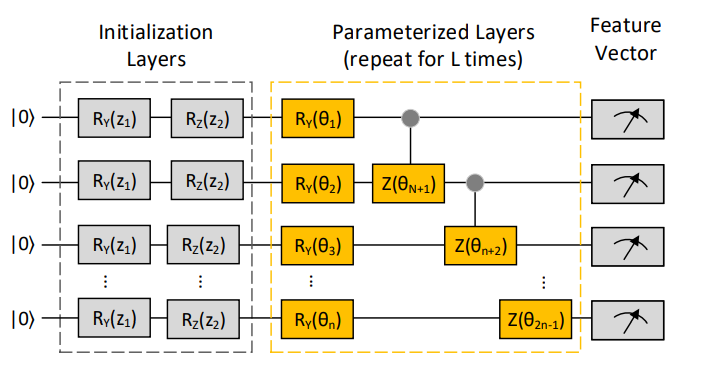
QGAN-HG Neural Network
After generating a feature vector from the QGAN-HG circuit, these vectors are then fed to the classic neural network. The output of this neural network contains atom and bond layers used to generate atom vectors and bond matrices.
Metrics used for calculation
- Frechet Distance: It measures the similarity between real and synthetic molecule distribution.
- Drug Properties: Drug Properties include Drug Likeliness, Solubility, Synthesizability. Together with other properties, they are measured using RDKit.
Model Architecture
As shown in the figure above:

- The first image consists of drug fragments and binding site(receptor). If the shape and pose of the drug fit into the receptor, it means it can cure the disease. This is the fundamental phenomenon and can be explained with the lock and key concept where lock is the receptor and key acts as a drug.
- The second image consists of two image quantum stage and classical stage separated by dotted line.
- The third image consists of an atom and bond matrix to make a graphical structure of a synthetic molecule.
- In the final step, real molecules and synthetic molecules(from step 3) are fed into the classical discriminator for discriminating between the two and Frechet distance and drug properties are evaluated using the RDKit package. The final prediction is being propagated to two neural networks and quantum circuits so as to update all the parameters in each training epoch.
Dataset used to train the model
The dataset used for training this model was QM9 dataset which consists of 134K stable small organic molecules with up to nine heavy atoms.
Implementation Details
The implementation of QGAN-HG is based on MolGAN, and its source code is dependent on:
Dependencies
This model depends on following framework:
- python>=3.5
- pytorch>=0.4.1: https://pytorch.org
- rdkit: https://www.rdkit.org
- pennylane
- tensorflow==1.15
- frechetdist
Demo of Pre-trained model of QGAN-HG
- Clone the Github repository and install all the required libraries. The full code snippet is available here.
!git clone https://github.com/jundeli/quantum-gan.git
- Change the directory, and run the bash commands to download all the datasets.
Command to change the directory is undermentioned.
import os
os.chdir("/content/quantum-gan/data/") Bash commands to download the datasets via .sh file is mentioned below.
%%bash
chmod u+x download_dataset.sh
./download_dataset.sh
- Then run the script to convert the downloaded dataset in a graph format.
!python sparse_molecular_dataset.py
- Again change the directory to the cloned repository to access the pre-trained models.
os.chdir("/content/quantum-gan/")
- Import all the required libraries and packages. The code snippet is available here.
- Set up your qubit units and generate a quantum circuit(discussed above).
Initialize two random uniform noise parameters z1 and z2.
# random noise as generator input
#z1 and z2 are random noise parameters.
z1 = random.uniform(-1, 1)
z2 = random.uniform(-1, 1)Then generate a circuit for both the atom vector and node matrix(as shown in figure above). More details of how to create a quantum circuit via Pennylane is available here.
@qml.qnode(dev, interface='torch')
def gen_circuit(w):
# random noise as generator input
#z1 and z2 are random noise parameters.
z1 = random.uniform(-1, 1)
z2 = random.uniform(-1, 1)
#number of layers in the circuit
layers = 1
# construct generator circuit for both atom vector and node matrix
for i in range(qubits):
qml.RY(np.arcsin(z1), wires=i)
qml.RZ(np.arcsin(z2), wires=i)
for l in range(layers):
for i in range(qubits):
qml.RY(w[i], wires=i)
for i in range(qubits-1):
qml.CNOT(wires=[i, i+1])
qml.RZ(w[i+qubits], wires=i+1)
qml.CNOT(wires=[i, i+1])
return [qml.expval(qml.PauliZ(i)) for i in range(qubits)]- Now, create an argument parser and pass all the parameters as arguments. The parameters are model configuration, training configuration, Quantum circuit configuration, step size(number of iterations, learning rate, etc). The code snippet is available here.
- Now, initialize the pre-trained QGAN-HG model. The code of it is shown below. The config represents a dictionary of all the parameter initialized above in Step 7
self = Solver(config)
- Next, is to generate the inference from the pre-trained model and calculate the loss for synthetic and real molecules.
# Start inference.
print('Start inference...')
start_time = time.time()
#here a is the adjacency matrix and
#x are the nodes
mols, _, _, a, x, _, _, _, _ = self.data.next_train_batch(self.batch_size)
a = torch.from_numpy(a).to(self.device).long() # Adjacency.
x = torch.from_numpy(x).to(self.device).long() # Nodes.
#"Convert label indices to one-hot vectors
a_tensor = self.label2onehot(a, self.b_dim)
x_tensor = self.label2onehot(x, self.m_dim)
z = torch.stack(tuple(ibm_sample_list)).to(self.device).float()
# Z-to-target
# # Compute loss with fake images.
edges_logits, nodes_logits = self.G(z)
# Postprocess with Gumbel softmax
(edges_hat, nodes_hat) = self.postprocess((edges_logits, nodes_logits), self.post_method)
logits_fake, features_fake = self.D(edges_hat, None, nodes_hat)
g_loss_fake = - torch.mean(logits_fake)
#compute the loss with real images
# Real Reward
rewardR = torch.from_numpy(self.reward(mols)).to(self.device)
# Fake Reward
(edges_hard, nodes_hard) = self.postprocess((edges_logits, nodes_logits), 'hard_gumbel')
edges_hard, nodes_hard = torch.max(edges_hard, -1)[1], torch.max(nodes_hard, -1)[1]
mols = [self.data.matrices2mol(n_.data.cpu().numpy(), e_.data.cpu().numpy(), strict=True)
for e_, n_ in zip(edges_hard, nodes_hard)]
rewardF = torch.from_numpy(self.reward(mols)).to(self.device)
# Value loss
value_logit_real,_ = self.V(a_tensor, None, x_tensor, torch.sigmoid)
value_logit_fake,_ = self.V(edges_hat, None, nodes_hat, torch.sigmoid)
g_loss_value = torch.mean((value_logit_real - rewardR) ** 2 + (
value_logit_fake - rewardF) ** 2)
R=[list(a[i].reshape(-1)) for i in range(self.batch_size)]
F=[list(edges_hard[i].reshape(-1)) for i in range(self.batch_size)]
fd_bond_only = frdist(R, F)
R=[list(x[i]) + list(a[i].reshape(-1)) for i in range(self.batch_size)]
F=[list(nodes_hard[i]) + list(edges_hard[i].reshape(-1)) for i in range(self.batch_size)]
fd_bond_atom = frdist(R, F)
loss = {}
loss['G/loss_fake'] = g_loss_fake.item()
loss['G/loss_value'] = g_loss_value.item()
loss['FD/fd_bond_only'] = fd_bond_only
loss['FD/fd_bond_atom'] = fd_bond_atom- The last step is to evaluate all the molecules by RDKit. The code for it is available here and the output of it is shown below.

You can check the full demo here.
Conclusion
In this article, we have discussed Quantum GAN with Hybrid Generator. The advantages of using this model are listed below:
- Qubit-friendly requires less number of the qubit to perform quantum computation.
- High training efficiency.
- Generate good molecular graphs given the Frechet distance and drug properties.
Official code, Docs & Tutorial are available at: